Very nice 20 min presentation to non-specialists by computational chemist Marcel Swart.
Thursday, December 27, 2012
Marcel Swart - I am a (computational) chemist
Very nice 20 min presentation to non-specialists by computational chemist Marcel Swart.
Wednesday, December 19, 2012
Molecular Modeling Basics Reviewed in Journal of Chemical Education

Docking made easy

Sunday, December 16, 2012
Conformational and rotational entropy and point group symmetry
The point group affects the free energy
The use of point group symmetry in quantum chemical calculations can speed up calculations significantly, but it is often difficult to input symmetric coordinates correctly so many people opt to run calculations without symmetry (i.e. in $C_1$ symmetry) anyway. However, the lack of symmetry changes the rotational entropy and, hence, the free energy you compute. So if you have symmetric molecules but choose to run in $C_1$ symmetry you must correct the entropies and free energies.
The rotational entropy is given by$$S_{rot}=R\ln\left(\frac{8\pi^2}{\sigma}\left(\frac{2\pi ekT}{h^2}\right)^{3/2}\sqrt{I_1I_2I_3}\right)$$ $\sigma$ is called the symmetry number and depends on the point group: for example, $\sigma=1$ for $C_1$ and $C_s$, $\sigma=n$ for $C_{nv}$ and $C_{nh}$, $\sigma= 2n$ for $D_{nh}$ and $D_{nd}$, and $\sigma= 12$ or $T_d$. So the rotational entropy calculated with and without symmetry will differ by $R\ln(\sigma)$:$$S_{rot}=S_{rot}^{C_1}-R\ln(\sigma)$$and similarly for the free energy$$G^\circ=G^{\circ,C_1}+RT\ln(\sigma)$$
An example: $H_2O+Cl^-\rightleftharpoons HOH\cdots Cl^-$
The free energy change for this reaction is$$\Delta G^\circ=\Delta G^{\circ,C_1}+RT\ln\left(\frac{\sigma_{HOH\cdots Cl^-(C_s)}}{\sigma_{H_2O(C_{2v})}\sigma_{Cl^-(C_1)}}\right)\\ \Delta G^\circ=\Delta G^{\circ,C_1}+RT\ln\left(\frac{1}{2\times 1}\right)=\Delta G^{\circ,C_1}-RT\ln (2)$$The corresponding equilibrium constants are$$K=e^{-\Delta G^\circ/RT}=2K^{C_1}$$So, $\sigma$'s accounts for the fact that, because of the symmetry of water, there are two ways of making $HOH\cdots Cl^-$and another way to view $R\ln(2)$ is that it is the conformational entropy of the complex.
A test: $H_2O+NH_3\rightleftharpoons HOH\cdots NH_3$
For the above equilibrium what is $X$ in$$\Delta G^\circ=\Delta G^{\circ,C_1}-RT\ln (X)$$
An exception: $2H_2O\rightleftharpoons HOH\cdots OH_2$
Based on the rules outlined so far one would expect the free energy change for this equilibrium to be$$\Delta G^\circ=\Delta G^{\circ,C_1}-RT\ln (4)$$The factor of four accounts for the fact that there are four ways of making $HO^AH\cdots O^BH_2$. However, since the water molecules are identical there are actually four additional water dimer possibilities for $HO^BH\cdots O^AH_2$, so$$\Delta G^\circ=\Delta G^{\circ,C_1}-RT\ln (8)$$In general, for $A+A\rightleftharpoons Product$ reactions the symmetry number for $A+A$ is $2\sigma_A^2$rather than $\sigma_A^2$
All these considerations also apply to activation free energies and rate constants as outlined in this excellent paper by Fernández-Ramos et al., which inspired this post. See also this excellent paper by Gilson and Irikura.

This work is licensed under a Creative Commons Attribution 3.0 Unported License.
Wednesday, December 12, 2012
Thermodynamics in solution: a brief guide for quantum chemists
2015.01.25: Please read this paper instead of this post. It turns out most of what I write here is wrong.
The thermodynamic properties such as enthalpy, entropy, and free energy you get from a vibrational analysis by programs such as GAMESS and Gaussian are those of an ideal gas at 1 bar pressure (and usually 298 K). If you calculate free energy changes in solution (using methods such as PCM or COSMO) there are a few things you should do differently compared to gas phase calculations. These things will only make a difference if you are computing free energy changes for processes where the number of particles change, such as binding free energies. The corrections will cancel out for things such as conformational free energy differences.
Use Helmholtz free energies instead of Gibbs free energies
Experimental studies typically report Gibbs free energy changes ($\Delta G^\circ$), which are related to Helmholtz free energy changes ($\Delta A^\circ$) by$$\Delta G^\circ = \Delta A^\circ + p^\circ \Delta V$$ $\Delta V$ is the change in volume of the solution due to the reaction. This volume change is negligible so $\Delta G^\circ = \Delta A^\circ$ is a good approximation.
The Gibbs free energy printed by quantum chemistry program correspond to an ideal gas where $pV=RT$ and the difference between $\Delta G^\circ$ and $\Delta A^\circ$ is much larger.
Computing free energies with semi-empirical methods
Most semiempirical methods such as AM1 and PM6 are parameterized such that the electronic energy matches experimental heat of formations ($\Delta H_f^\circ$) at 298 K. So a gas phase free energy change should be computed as$$\Delta G^\circ=\Delta\Delta H_f^\circ-T\Delta S^\circ$$i.e. you don't need any of the enthalpy information printed out as part of the vibrational analysis. A solution free energy change should be computed as $$\Delta A^\circ=\Delta\Delta U_f-T\Delta S^\circ$$where $\Delta U_f=\Delta H_f^\circ-RT$
Added 2013.08.16: However, dispersion and/or hydrogen bond corrected semi-empirical methods such as PM6-DH+ are parameterized against electronic binding energies. So if you are computing binding free energies with such methods you need to add the translational, rotational, and vibrational enthalpy corrections to $\Delta\Delta H_f^\circ$.
Change the standard state to 1 mol per liter
The translational entropy printed out depends on the volume of the system, which is computed as $V=RT/p$ = 24.79 liters. For solution that volume should be 1 liter. Most programs do not allow you to change volume so you must apply the correction to the entropy$$S_{soln}^\circ=S_{gas}^\circ+R\ln\left(\frac{1}{24.79}\right)$$ yourself
Is it OK to use the rigid rotor-harmonic oscillator approximation in solution?
Short answer: yes. The derivations of the translational, rotational, and vibrational free energies are done for an isolated molecule in vacuum, while a molecule in solution interacts with solvent molecules. However, a molecule in solution still has the exact same degrees of freedom as in the gas phase: it is free to explore the entire three dimensional volume and is free to adapt any rotational angle just like in the gas phase, so the resulting free energy expressions are the same. Similarly, the individual molecules still have $3N-6$ internal vibrational frequencies. The energy from the solute-solvent vibrations are included in the solvation free energy.
This blogpost was inspired by this excellent paper by Muddana and Gilson, and I thank Mike Gilson for helpful discussions.
This work is licensed under a Creative Commons Attribution 3.0 Unported License.
Saturday, December 1, 2012
Computational Chemistry Highlights: November issue
The November issue of Computational Chemistry Highlights is out.
CCH is an overlay journal that identifies the most important papers in computational and theoretical chemistry published in the last 1-2 years. CCH is not affiliated with any publisher: it is a free resource run by scientists for scientists. You can read more about it here.
Table of content for this issue features contributions from CCH editors Steven Bachrach, Marcel Swart, Thomas Cundari, and Jan Jensen:
A Hierarchy of Methods for the Energetically Accurate Modeling of Isomerism in monosaccarides
CCH is an overlay journal that identifies the most important papers in computational and theoretical chemistry published in the last 1-2 years. CCH is not affiliated with any publisher: it is a free resource run by scientists for scientists. You can read more about it here.
Table of content for this issue features contributions from CCH editors Steven Bachrach, Marcel Swart, Thomas Cundari, and Jan Jensen:
A Hierarchy of Methods for the Energetically Accurate Modeling of Isomerism in monosaccarides
Saturday, November 17, 2012
Building molecules using fragments in Avogadro
A new version (1.1.0) of Avogadro was released recently and contains a very useful new building tool, where molecule-fragments can be added to selected atoms. The screencast shows how.
Remember: please cite the Avogadro paper if you use Avogadro in research that leads to a publication
This work is licensed under a Creative Commons Attribution 3.0
Thursday, November 1, 2012
Computational Chemistry Highlights: October issue
The October issue of Computational Chemistry Highlights is out.
CCH is an overlay journal that identifies the most important papers in computational and theoretical chemistry published in the last 1-2 years. CCH is not affiliated with any publisher: it is a free resource run by scientists for scientists. You can read more about it here.
Table of content for this issue features contributions from CCH editors Steven Bachrach, Grant Hill, and Jan Jensen:
DSD-DFT – a double hybrid variation
Palladium-Catalyzed C–H Activation Taken to the Limit. Flattening an Aromatic Bowl by Total Arylation
Empirical correction of nondynamical correlation energy for density functionals
Variational approach for nonpolar solvation analysis
Why a Proximity-Induced Diels–Alder Reaction Is So Fast
Interested in more? There are many ways to subscribe to CCH updates.
CCH is an overlay journal that identifies the most important papers in computational and theoretical chemistry published in the last 1-2 years. CCH is not affiliated with any publisher: it is a free resource run by scientists for scientists. You can read more about it here.
Table of content for this issue features contributions from CCH editors Steven Bachrach, Grant Hill, and Jan Jensen:
DSD-DFT – a double hybrid variation
Palladium-Catalyzed C–H Activation Taken to the Limit. Flattening an Aromatic Bowl by Total Arylation
Empirical correction of nondynamical correlation energy for density functionals
Variational approach for nonpolar solvation analysis
Why a Proximity-Induced Diels–Alder Reaction Is So Fast
Interested in more? There are many ways to subscribe to CCH updates.
Saturday, October 20, 2012
Video introductions to GaussView/Gaussian09
Video's made by Jeff Yarger at Arizona State University
Introduction to Electronic Structure Calculation Software
First Example of Building Molecule in Gaussview
Example of Thermochemistry Calculation in Gaussian 09
Introduction to Electronic Structure Calculation Software
First Example of Building Molecule in Gaussview
Example of Thermochemistry Calculation in Gaussian 09
Wednesday, October 3, 2012
Peer instruction questions for DFT
Readers of this blog will know I am a fan of using peer instruction in teaching and I am happy to say that, as of this week, I am using peer instruction instruction in all the courses I contribute to. This week I covered DFT in our Computational Chemistry course (which is the only week I currently contribute).
In the past I have always lectured, straight from the DFT section in my book, which the students get a copy of beforehand; all because I simply couldn't think of good multiple choice questions. But now I use Socrative for polling, which has a very nifty "short answer" option: I ask a question, the students discuss and type in their answer, once finished they then get to vote for their favorite answer, and, once finished, I discuss some of the answers (including why some answers are wrong). You can see the questions I asked above.
Of course this means the students have to prepare for class beforehand. I had them read the electron correlation and DFT sections from my book, a perspective article by Kieron Burke, and the DFT chapter from Frank Jensen's book. In addition they had to watch David Sherrill's video lecture on DFT:
I am very happy with the way the process went. I'll definitely do it this way from now on.
Computational Chemistry Highlights: September issue
The September issue of Computational Chemistry Highlights is out.
CCH is an overlay journal that identifies the most important papers in computational and theoretical chemistry published in the last 1-2 years. CCH is not affiliated with any publisher: it is a free resource run by scientists for scientists. You can read more about it here.
Table of content for this issue features contributions from CCH editors Steven Bachrach, G. Andres Cisneros, and Jan Jensen:
CCH is an overlay journal that identifies the most important papers in computational and theoretical chemistry published in the last 1-2 years. CCH is not affiliated with any publisher: it is a free resource run by scientists for scientists. You can read more about it here.
Table of content for this issue features contributions from CCH editors Steven Bachrach, G. Andres Cisneros, and Jan Jensen:
Saturday, September 29, 2012
Georgia Tech's Summer Lectures Series in Theoretical Chemistry
I just came across this set of lectures in theoretical chemistry:
"The following lectures were presented as part of Georgia Tech's Summer Lectures Series in Theoretical Chemistry, for new graduate students and for participants in Georgia Tech's Research Experiences for Undergraduates (REU) program sponsored by the National Science Foundation. These lectures focus on electronic structure theory and were prepared by the Sherrill research group. The lectures are being made publicly available in the interests of enhancing education in theoretical and computational chemistry."
Sunday, September 23, 2012
Video lectures on basis sets and potential energy surfaces by Luca De Vico
Video lectures by Luca De Vico:
Basis sets: an introduction
Optimization techniques: an introduction - part 1
Optimization techniques: an introduction - part 2
Optimization techniques: an introduction - part 3
Basis sets: an introduction
Optimization techniques: an introduction - part 1
Optimization techniques: an introduction - part 2
Optimization techniques: an introduction - part 3
Thursday, September 6, 2012
Peer instruction questions for very basic molecular quantum mechanics
As I mentioned in my previous blog post I this year I made some screencasts of lectures to free up time for peer instruction questions. Here are the peer instruction questions I used.
This time I tried Socrative for voting, which allows students to type in short answers to questions. I can then select some of the questions and have students vote on their favorite. This is what's happening in slide 13 and 23.
You'll notice I also made use of Molecule Calculator, which is introduced here. Next year I have to remember also to assign the MolCalc intro video.
This work is licensed under a Creative Commons Attribution 3.0 Unported License.
This time I tried Socrative for voting, which allows students to type in short answers to questions. I can then select some of the questions and have students vote on their favorite. This is what's happening in slide 13 and 23.
You'll notice I also made use of Molecule Calculator, which is introduced here. Next year I have to remember also to assign the MolCalc intro video.
This work is licensed under a Creative Commons Attribution 3.0 Unported License.
Saturday, September 1, 2012
Very quick introduction to molecular quantum mechanics for simple systems
My guest lecture in the course Unifying Concepts in Nano-Science back in 2009 somehow turned into a regular gig. This year I made a few more screencasts of parts of my lecture to free up time for peer instruction questions during the lecture period:
a. A brief motivation for the Schrödinger equation
b. Four simple examples of the Schrödinger equation
c. Solutions to the Schrödinger equation for three simple systems
d. Changes in quantum states and spectroscopy: a simple example
e. Tunneling and scanning tunneling microscopy
The simulations are done using a free program called Molecular Workbench. Once you have installed Molecular Workbench you can download the list of simulations here.
2012.09.07 Update: The slides used in the video and the peer instruction questions can be found here
This work is licensed under a Creative Commons Attribution 3.0 Unported License.
b. Four simple examples of the Schrödinger equation
c. Solutions to the Schrödinger equation for three simple systems
d. Changes in quantum states and spectroscopy: a simple example
e. Tunneling and scanning tunneling microscopy
The simulations are done using a free program called Molecular Workbench. Once you have installed Molecular Workbench you can download the list of simulations here.
2012.09.07 Update: The slides used in the video and the peer instruction questions can be found here
This work is licensed under a Creative Commons Attribution 3.0 Unported License.
Computational Chemistry Highlights: August issue
The August issue of Computational Chemistry Highlights is out.
CCH is an overlay journal that identifies the most important papers in computational and theoretical chemistry published in the last 1-2 years. CCH is not affiliated with any publisher: it is a free resource run by scientists for scientists. You can read more about it here.
Table of content for this issue features contributions from CCH editors Steven Bachrach, Victor Guallar, and Jan Jensen:
Effect of Hydrogen Bonds on pKa Values: Importance of Networking
CCH is an overlay journal that identifies the most important papers in computational and theoretical chemistry published in the last 1-2 years. CCH is not affiliated with any publisher: it is a free resource run by scientists for scientists. You can read more about it here.
Table of content for this issue features contributions from CCH editors Steven Bachrach, Victor Guallar, and Jan Jensen:
Effect of Hydrogen Bonds on pKa Values: Importance of Networking
Wednesday, August 15, 2012
Avogadro: An advanced semantic chemical editor, visualization, and analysis platform
 |
| from: http://avogadro.openmolecules.net/wiki/File:Avogadro-unitcell.png |
1) The paper probably describes options in Avogadro you didn't know about, so check it out.
2) Please support the Avogadro developers by citing this paper when you are writing up projects where you have used Avogadro in some way.
This also seems a good time to say Thank You! to Geoff, Marcus, Donald, David, Tim, and Eva for all their hard work and, not least, for making it freely available to the rest of us.
This work is licensed under a Creative Commons Attribution 3.0
Tuesday, August 14, 2012
The Molecule Calculator
What is MolCalc?
MolCalc is a web interface that allows anyone to build (small*) molecules and estimate** molecular properties such as molecular structure, heats of formation, vibrational frequencies and vibrational modes, and molecular orbitals and orbital energies in a matter of seconds or minutes - depending on the size.
MolCalc is designed for teaching as opposed to research - specifically for assignments in which students build their own molecules and estimate their own molecular properties. (**MolCalc is therefore designed to run fast and the estimated molecular properties will not match experimental values exactly, and in some cases be quite different.) The idea is to have students develop a “chemical intuition” about how molecular structure affects molecular properties, without performing the underlying calculations by hand (which would be near impossible for all but the simplest chemical systems).
How can I use MolCalc in teaching?
Just like a pocket calculator or a symbolic math program (such as Mathematica or MAPLE), MolCalc allows one to assign “higher level” chemical problems that are not practically possible to solve otherwise.
For example, one might now ask students to compute the effect of a substituent on a particular vibration, and then rationalize the effect using molecular orbitals. Or one might ask more open ended questions such as “build a molecule with an unusually long C-C single bond”.
How does MolCalc work?
In the Molecule Editor page the molecular structure is build using Jmol and energy minimized using the UFF force field as implemented in Jmol.
In the Molecule Calculator page the structure is re-optimized at the PM3 level of theory for a maximum of 50 steps. This structure is then used to compute the heat of formation or vibrational frequencies at the PM3 level of theory, or the molecular orbitals using the RHF/STO-3G level of theory. These calculations are performed with the GAMESS program. OpenBabel is used to manage input files and coordinate files.
*MolCalc 1.0 allows calculations on (closed shell) molecules with only doubly occupied molecular orbitals and with less than 11 non-hydrogen atoms.
Can I modify and/or install MolCalc on my own server?
Yes, MolCalc is distributed through github under the GPL license (
). You must obtain a copy of the GAMESS code separately from http://www.msg.ameslab.gov/gamess/download.html
The interface code uses PHP5, jQuery, HTML5, and CSS3 and is very modular. It therefore quite easy to add new capabilities to MolCalc.
What if I find a bug in MolCalc?
Please report it here.
Who is involved with MolCalc?
MolCalc 1.0 is written by Jimmy Charnley Kromann based on an idea by Jan Jensen. Toke Fritzemeier wrote an early prototype. The Molecule Editor was inspired, in part, by the Virtual Molecular Modeling Kit.
The development of MolCalc is supported by the University of Copenhagen through the Education at its Best initiative (Den gode uddannelse).
This work is licensed under a Creative Commons Attribution 3.0 Unported License.
Sunday, August 12, 2012
Perspective on Density Functional Theory by Kieron Burke

Kieron Burke has written a very readable perspective on density functional theory in Journal of Chemical Physics, who, very commendably, has made it freely accessible.
To whet your appetite here are some quotes from the paper:
If you enjoyed the article you may also want to check out this presentation of Kieron's entitled "Density Functional Theory: A great physics success story"
If you wish to annoy and confuse a traditional quantum chemist, ask “How much correlation is there in the KS wavefunction?”
When teaching chemistry students, I explain that DFT is some algorithm meaning unreliable, while ab initio is Latin for too expensive.
Interestingly, I have recently co-authored an approximation with about 105 “empirical” parameters.111 Don Truhlar, eat your heart out!
Users should stick to the standard functionals (as most do, according to Fig. 1), or explain very carefully why not.
Thanks to Sten Rettrup for alerting me this paper
Saturday, August 4, 2012
Computational Chemistry Highlights: July Issue
The July issue of Computational Chemistry Highlights is out.
CCH is an overlay journal that identifies the most important papers in computational and theoretical chemistry published in the last 1-2 years. CCH is not affiliated with any publisher: it is a free resource run by scientists for scientists. You can read more about it here.
Table of content for this issue features contributions from CCH editors Steven Bachrach, Grant Hill, and Robert Paton:
Aromatic Transition States in Nonpericyclic Reactions: Anionic 5 -Endo Cyclylizations are Aborted Sigmatropic Shifts

CCH is an overlay journal that identifies the most important papers in computational and theoretical chemistry published in the last 1-2 years. CCH is not affiliated with any publisher: it is a free resource run by scientists for scientists. You can read more about it here.
Table of content for this issue features contributions from CCH editors Steven Bachrach, Grant Hill, and Robert Paton:
Aromatic Transition States in Nonpericyclic Reactions: Anionic 5 -Endo Cyclylizations are Aborted Sigmatropic Shifts

Monday, July 2, 2012
Computational Chemistry Highlights: June issue
The June issue of Computational Chemistry Highlights is out.
CCH is an overlay journal that identifies the most important papers in computational and theoretical chemistry published in the last 1-2 years. CCH is not affiliated with any publisher: it is a free resource run by scientists for scientists. You can read more about it here.
Table of content for this issue features contributions from CCH editors Steven Bachrach, Grant Hill, and Jan Jensen:
Interested in more? There are many ways to subscribe to CCH updates.
CCH is an overlay journal that identifies the most important papers in computational and theoretical chemistry published in the last 1-2 years. CCH is not affiliated with any publisher: it is a free resource run by scientists for scientists. You can read more about it here.
Table of content for this issue features contributions from CCH editors Steven Bachrach, Grant Hill, and Jan Jensen:
Monday, June 25, 2012
Teaching computational chemistry with the ADF program

Tuesday, June 5, 2012
Complete basis set limit extrapolation
The complete basis set (CBS) limit is not a basis set though it is often written as such, e.g. B3LYP/CBS. Instead the CBS limit is an extrapolated estimate of a result obtained using an infinitely large (complete) basis set. In principle this procedure removes any error due to the linear combination of atomic orbitals approximation, and any remaining disagreement with experiment is due to some other approximation such as the treatment of correlation. For many properties the CCSD(T)/CBS value can be regarded as a numerically exact for all practical purposes, i.e. it is unlikely that any higher level of theory predict significantly better results.
The extrapolation is based on a minimum of three separate calculations with increasingly larger basis sets. CBS limit extrapolation works only with basis sets designed specifically for the task, such as the correlation- or polarization-consistent basis sets, e.g. cc-pVxZ or pc-n.
The procedure is as follows: a given property $Y$ of interest (e.g. a relative energy, a frequency, or a bond length) is computed at a given level of theory (e.g. B3LYP) using at least three basis sets (e.g. cc-VDZ, cc-VTZ, and cc-VQZ. These data points a then fit to an equation, the two most popular equations are given here
Here, $Y_{CBS}$ is the CBS limit we're after and $x$ is 2 for cc-pVDZ, 3 for cc-pVTZ, and so on. $x$ is also often written as $L_{max}$ (or $l_{max}$), which is the highest angular momentum included in the basis set. For cc-pVDZ this means $d$ orbitals, which have an angular momentum of 2, so $x$ and $L_{max}$ are really the same.
Equation (1) contains three parameters ($Y_{CBS}$, $A$, and $B$) so a minimum of three different basis sets are needed to determine them. While Equation (2) only has two parameters, a minimum of three data points are still needed for reliable results.
For some properties and correlation methods the use of the double-zeta basis set does not lead to a good fit, so calculations with pentuple-zeta basis sets are necessary. There is some evidence that the pc-n basis set provides faster convergence to the CMS limit. CBS limit extrapolation is computationally very demanding and is typically done on relatively small systems to provide benchmark values to test more efficient methods.
Acknowledgment: I thank Anders Christensen for providing me with key papers and with helpful discussion.
This work is licensed under a Creative Commons Attribution 3.0
The extrapolation is based on a minimum of three separate calculations with increasingly larger basis sets. CBS limit extrapolation works only with basis sets designed specifically for the task, such as the correlation- or polarization-consistent basis sets, e.g. cc-pVxZ or pc-n.
The procedure is as follows: a given property $Y$ of interest (e.g. a relative energy, a frequency, or a bond length) is computed at a given level of theory (e.g. B3LYP) using at least three basis sets (e.g. cc-VDZ, cc-VTZ, and cc-VQZ. These data points a then fit to an equation, the two most popular equations are given here
$Y(x)=Y_{CBS}+Ae^{-Bx}$ (1)
$Y(x)=Y_{CBS}+Ax^{-3}$ (2)
Here, $Y_{CBS}$ is the CBS limit we're after and $x$ is 2 for cc-pVDZ, 3 for cc-pVTZ, and so on. $x$ is also often written as $L_{max}$ (or $l_{max}$), which is the highest angular momentum included in the basis set. For cc-pVDZ this means $d$ orbitals, which have an angular momentum of 2, so $x$ and $L_{max}$ are really the same.
Equation (1) contains three parameters ($Y_{CBS}$, $A$, and $B$) so a minimum of three different basis sets are needed to determine them. While Equation (2) only has two parameters, a minimum of three data points are still needed for reliable results.
For some properties and correlation methods the use of the double-zeta basis set does not lead to a good fit, so calculations with pentuple-zeta basis sets are necessary. There is some evidence that the pc-n basis set provides faster convergence to the CMS limit. CBS limit extrapolation is computationally very demanding and is typically done on relatively small systems to provide benchmark values to test more efficient methods.
Acknowledgment: I thank Anders Christensen for providing me with key papers and with helpful discussion.
This work is licensed under a Creative Commons Attribution 3.0
Friday, June 1, 2012
Computational Chemistry Highlights: May issue
The May issue of Computational Chemistry Highlights is out.
CCH is an overlay journal that identifies the most important papers in computational and theoretical chemistry published in the last 1-2 years. CCH is not affiliated with any publisher: it is a free resource run by scientists for scientists. You can read more about it here.
Table of content for this issue features contributions from CCH editors Steven Bachrach, Dean Tantillo, Dmitri Fedorov, and Jan Jensen:
Computational Design and Selection of Optimal Organic Photovoltaic Materials
Energy Decomposition Analysis in Solution Based on the Fragment Molecular Orbital Method
Amino acid-catalyzed aldol and Michael reactions
ROBIA and Dolabriferol
Quantitative NMR-Derived Interproton Distances Combined with Quantum Mechanical Calculations of 13C Chemical Shifts in the Stereochemical Determination of Conicasterol F, a Nuclear Receptor Ligand from Theonella swinhoei
Interested in more? There are many ways to subscribe to CCH updates.
Thursday, May 3, 2012
Lectures in electronic structure theory by Jack Simons
Wonderful! Here is the first lecture. You can find the rest here.
Tuesday, May 1, 2012
Computational Chemistry Highlights: April issue
The April issue of Computational Chemistry Highlights is out.
CCH is an overlay journal that identifies the most important papers in computational and theoretical chemistry published in the last 1-2 years. CCH is not affiliated with any publisher: it is a free resource run by scientists for scientists. You can read more about it here.
Table of content for this issue features contributions from CCH editors Steven Bachrach, Kieron Burke, Andres Cisneros, Sason Shaik, Robert Paton, Marcel Swart, and Jan Jensen:
Enzymatic catalysis of anti-Baldwin ring closure in Polyether Biosynthesis
Fast and Accurate Modeling of Molecular Atomization Energies with Machine Learning
Resolution of identity approach for the Kohn-Sham correlation energy with exact-exchange random-phase approximation
Steric Crowding Can Stabilize a Labile Molecule: Solving the Hexaphenyethane Riddle
Hydrogen-bond stabilization in oxyanion holes: grand jetè to three dimensions
Conformations and Fluorescence of Encapsulated Stilbene
Quadruple bonding in C2 and analogous eight-valence electron species
Exchange-Enhanced Open-Shell States: Hund's rule for Bioinorganic Species Applied to H-abstraction
Editorial: First indication of impact
Interested in more? There are many ways to subscribe to CCH updates.
CCH is an overlay journal that identifies the most important papers in computational and theoretical chemistry published in the last 1-2 years. CCH is not affiliated with any publisher: it is a free resource run by scientists for scientists. You can read more about it here.
Table of content for this issue features contributions from CCH editors Steven Bachrach, Kieron Burke, Andres Cisneros, Sason Shaik, Robert Paton, Marcel Swart, and Jan Jensen:
Enzymatic catalysis of anti-Baldwin ring closure in Polyether Biosynthesis
Fast and Accurate Modeling of Molecular Atomization Energies with Machine Learning
Resolution of identity approach for the Kohn-Sham correlation energy with exact-exchange random-phase approximation
Steric Crowding Can Stabilize a Labile Molecule: Solving the Hexaphenyethane Riddle
Hydrogen-bond stabilization in oxyanion holes: grand jetè to three dimensions
Conformations and Fluorescence of Encapsulated Stilbene
Quadruple bonding in C2 and analogous eight-valence electron species
Exchange-Enhanced Open-Shell States: Hund's rule for Bioinorganic Species Applied to H-abstraction
Editorial: First indication of impact
Interested in more? There are many ways to subscribe to CCH updates.
Sunday, April 15, 2012
Virtual Molecular Modeling Kit
I recently came across a very interesting resource called the Virtual Molecular Modeling Kit. It looks like it can do an awful lots of things and I have only scratched the surface myself.
Very commendably, the authors Otis Rothenberger and Thomas Newton, have created a set of instructional videos. The video below highlights the fact Jmol 12.2 and later versions now have a molecular editor, which I somehow had completely missed.
These, and related sites such as this one and this one, are examples of what I call chemical calculators. In analogy with conventional calculators, they allow effortless computations for certain quantities that, before, we would have to laboriously compute by hand, or approximate, or memorize.
Students are no longer required to manually compute square roots or take logarithms. We now have these calculators that can compute dipole moments, electrostatic potentials and orbitals, with roughly the same effort it takes to use a calculator. Perhaps we should stop requiring students to memorize nomenclature and perform trivial computations and start teaching them how to solve more interesting chemical problems using these tools.
Monday, April 2, 2012
Computational Chemistry Highlights: March issue
The March issue of Computational Chemistry Highlights is out.
CCH is an overlay journal that identifies the most important papers in computational and theoretical chemistry published in the last 1-2 years. CCH is not affiliated with any publisher: it is a free resource run by scientists for scientists. You can read more about it here.
Table of content for the March issue featuring contributions from CCH editors Patrik Rydberg, Steven Bachrach, and Jan Jensen:
CCH is an overlay journal that identifies the most important papers in computational and theoretical chemistry published in the last 1-2 years. CCH is not affiliated with any publisher: it is a free resource run by scientists for scientists. You can read more about it here.
Table of content for the March issue featuring contributions from CCH editors Patrik Rydberg, Steven Bachrach, and Jan Jensen:
Interested in more? There are many ways to subscribe to CCH updates.
Sunday, March 25, 2012
Fourteen Easy Lessons in Density Functional Theory
John Perdew and Adrienn Ruzsinszky have published a fantastically readable summary of density functional theory in International Journal of Quantum Chemistry, who, very commendably, has made it freely available.

Tuesday, March 6, 2012
Computational Chemistry Highlights: recent important articles in molecular modeling
The first (February) issue of Computational Chemistry Highlights is out.
CCH is an overlay journal that identifies the most important papers in computational and theoretical chemistry published in the last 1-2 years. CCH is not affiliated with any publisher: it is a free resource run by scientists for scientists. You can read more about it here.
Table of content for the February issue featuring contributions from CCH editors Jan Jensen and Steven Bachrach:
Total Synthesis of Oxidized Welwitindolinones and (-)-N-Methylwelwitindolinone C Isonitrile
Roaming-mediated isomerization in the photodissociation of nitrobenzene
Assessment of Popular DFT and Semiempirical Molecular Orbital Techniques for Calculating Relative Transition State Energies and Kinetic Product Distributions in Enantioselective Organocatalytic Reactions
Regiochemical Substituent Switching of Spin States in Aryl(trifluoromethyl)carbenes
NMR Structure Determination for Larger Proteins Using Backbone-Only Data
Interested in more? There are already several contributions to the March issue and there are many ways to subscribe to updates.
CCH is an overlay journal that identifies the most important papers in computational and theoretical chemistry published in the last 1-2 years. CCH is not affiliated with any publisher: it is a free resource run by scientists for scientists. You can read more about it here.
Table of content for the February issue featuring contributions from CCH editors Jan Jensen and Steven Bachrach:
Total Synthesis of Oxidized Welwitindolinones and (-)-N-Methylwelwitindolinone C Isonitrile
Roaming-mediated isomerization in the photodissociation of nitrobenzene
Assessment of Popular DFT and Semiempirical Molecular Orbital Techniques for Calculating Relative Transition State Energies and Kinetic Product Distributions in Enantioselective Organocatalytic Reactions
Regiochemical Substituent Switching of Spin States in Aryl(trifluoromethyl)carbenes
NMR Structure Determination for Larger Proteins Using Backbone-Only Data
Interested in more? There are already several contributions to the March issue and there are many ways to subscribe to updates.
Sunday, February 26, 2012
A sense of scale 2
Another great site for getting a sense of scale. Frankly, shouldn't every chemistry lecture start with a reminder of what scale we are dealing with?
Related blog post
A sense of scale
Friday, February 24, 2012
Dancing orbitals
Amazing movie of dancing orbitals (aka charge transfer dynamics) from Felix over at Chemical Quantum Images.
Sunday, February 12, 2012
High density energy storage using self-assembled materials
This award winning movie was made by Chris Wilmer, a graduate student at the Snurr group at Northwesten. I am thoroughly impressed.
Via Wired.
Sunday, January 22, 2012
Negative activation energies in molecular modeling: diagnosis and cures
This post is inspired by a recent discussion with Paolo in the comments section of this post.
So, you've found that the energy of your transition state (TS) is lower than your reactants (i.e. you have a negative activation energy). Don't panic, instead pretend you're Dr House and go through it methodically:
Did you actually find the right TS?
A TS is a completely optimized geometry (no constraints, "zero" gradient) with one has one and only one imaginary frequency. Yes? OK, how big is the imaginary frequency and does the normal mode look like what you would expect? Sometimes minima can have small (< ca 100 cm-1) imaginary frequencies due to numerical "noise", or the TS search algorithm will find a TS for, say, methyl rotation. No, looks OK? (Consider computing an IRC do be really sure, just in case.) Let's move on.
How much lower?
Some processes have very low (< 1 kcal/mol) barriers. Numerical "noise" can cause these barriers to come out slightly negative (-0.1 to -1.0 kcal/mol) instead. If so, consider your reaction barrier-less for all intends and purposes. No, much more negative? Let's move on.
What energy are we talking about here?
If the activation energy is calculated using single point energies (for example B3LYP/6-31G(d)//RHF/3-21G) then the B3LYP/6-31G(d) PES may not have a barrier or the B3LYP/6-31G(d) TS geometry looks very different from the RHF/3-21G TS geometry.
The easiest way to check for this particular case is to geometry optimize your reactant at the B3LYP/6-31G(d) level of theory. If the optimization results in the product geometry, then there is (very likely) no barrier to the reaction. If the optimization gives you a reactant geometry, then one explanation is that 3-21G is not a reliable method for finding the TS and the solution is to use B3LYP/6-31G(d) in your TS search. There are other explanations, so read on.
If the activation energy is calculated using zero point energy or free energy corrections and is negative (this is not uncommon for reactions whose PES barriers are less than 2-3 kcal/mol) then you can consider the reaction barrier-less. If you want to be really sure, you can add these corrections along the entire IRC. However, there are also other explanations so read on.
Is your reaction unimolecular?
Is your reaction bimolecular?
OK, so you have found a reasonable-looking TS using some method and the electronic energy barrier computed using that method is quite negative. The first question is how did you compute the barrier?
If you computed the barrier as the energy difference between the TS and the bound complex of your two reactants (R1/R2) then the most likely explanation is that you have not found the lowest energy conformation of this complex (see the section on unimolecular reactions).
If you computed the barrier as the energy difference between the TS and the sum of the energies and your two isolated reactants [E(R1)+E(R2)] then there is not necessarily anything wrong with your calculations. Read on.
How should I compute the activation energy of a bimolecular reaction?
(This sub-section has been updated 2012.05.05)
The activation energy for a biomolecular reaction should be computed as
If this energy is negative then you would say that this is a barrier-less reaction and the reaction rate is proportional to the collision frequency.
So, you've found that the energy of your transition state (TS) is lower than your reactants (i.e. you have a negative activation energy). Don't panic, instead pretend you're Dr House and go through it methodically:
Did you actually find the right TS?
A TS is a completely optimized geometry (no constraints, "zero" gradient) with one has one and only one imaginary frequency. Yes? OK, how big is the imaginary frequency and does the normal mode look like what you would expect? Sometimes minima can have small (< ca 100 cm-1) imaginary frequencies due to numerical "noise", or the TS search algorithm will find a TS for, say, methyl rotation. No, looks OK? (Consider computing an IRC do be really sure, just in case.) Let's move on.
How much lower?
Some processes have very low (< 1 kcal/mol) barriers. Numerical "noise" can cause these barriers to come out slightly negative (-0.1 to -1.0 kcal/mol) instead. If so, consider your reaction barrier-less for all intends and purposes. No, much more negative? Let's move on.
What energy are we talking about here?
If the activation energy is calculated using single point energies (for example B3LYP/6-31G(d)//RHF/3-21G) then the B3LYP/6-31G(d) PES may not have a barrier or the B3LYP/6-31G(d) TS geometry looks very different from the RHF/3-21G TS geometry.
The easiest way to check for this particular case is to geometry optimize your reactant at the B3LYP/6-31G(d) level of theory. If the optimization results in the product geometry, then there is (very likely) no barrier to the reaction. If the optimization gives you a reactant geometry, then one explanation is that 3-21G is not a reliable method for finding the TS and the solution is to use B3LYP/6-31G(d) in your TS search. There are other explanations, so read on.
If the activation energy is calculated using zero point energy or free energy corrections and is negative (this is not uncommon for reactions whose PES barriers are less than 2-3 kcal/mol) then you can consider the reaction barrier-less. If you want to be really sure, you can add these corrections along the entire IRC. However, there are also other explanations so read on.
Is your reaction unimolecular?
$R \rightarrow TS \rightarrow P$
OK, so you have found a reasonable-looking TS using some method and the electronic energy barrier computed using that method is quite negative. If your reaction is unimolecular then the most likely explanation is that you have not found the lowest energy conformation of your reactant. If can't find a lower energy structure for your reaction, compute an IRC. An IRC follows the minimum energy path downhill to your reactant and product, so you will find structures with a lower energy than your TS.
Is your reaction bimolecular?
$R_1 + R_2 \rightarrow R_1/R_2 \rightarrow TS \rightarrow P$
If you computed the barrier as the energy difference between the TS and the bound complex of your two reactants (R1/R2) then the most likely explanation is that you have not found the lowest energy conformation of this complex (see the section on unimolecular reactions).
If you computed the barrier as the energy difference between the TS and the sum of the energies and your two isolated reactants [E(R1)+E(R2)] then there is not necessarily anything wrong with your calculations. Read on.
How should I compute the activation energy of a bimolecular reaction?
(This sub-section has been updated 2012.05.05)
The activation energy for a biomolecular reaction should be computed as
$E_a = E(TS) - [E(R_1) + E(R_2)]$
If this energy is negative then you would say that this is a barrier-less reaction and the reaction rate is proportional to the collision frequency.
Saturday, January 14, 2012
Sunday, January 8, 2012
The ideal app for an ideal gas

I recently came across a very cool app called Atoms in Motion ($2.99). With this app you can perform molecular dynamics simulations of He, Ne, Ar, and Kr (and mixtures thereof) at different temperatures, volumes, and number of atoms.
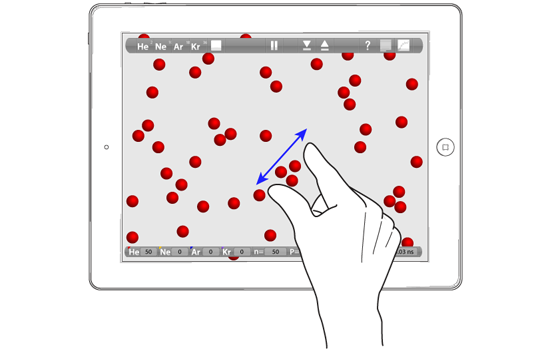
It is effortless to set up a simulation and change the conditions: swipe to change T (or shake the iPad!) and pinch to change V. You can also flick an atom and, frankly, the most fun is to build a few clusters at 1 K and then move an atom carefully in position and flick it at a cluster! Something I have dubbed "atomic billiards".
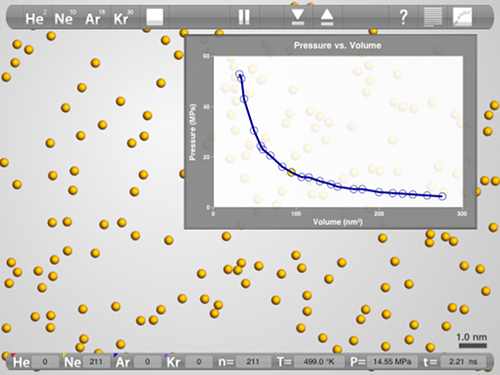
Teaching
Since not every student has an iPad (yet), the main teaching-use will probably be in lecture, projection the simulation on using a VGA adapter (hint: simulations make for great peer instruction questions). Because it is based on real simulations, and not pre-recorded movies, the app is very versatile and can be used to demonstrate very simple things like solids, liquids, and gases or an atomic view of temperature or relatively complex things like defects in solids and deviations from ideal gas behavior (using the plotting tool).
However, the ideal use of this app would really be self-guided discovery. Most features are very intuitive (the plotting tool takes some practice) and I'd love to see what a class of 10-year olds iPad users would get out of this. Side note: it would really have been great if the iPad could vibrate and the pressure could be linked to the vibration.
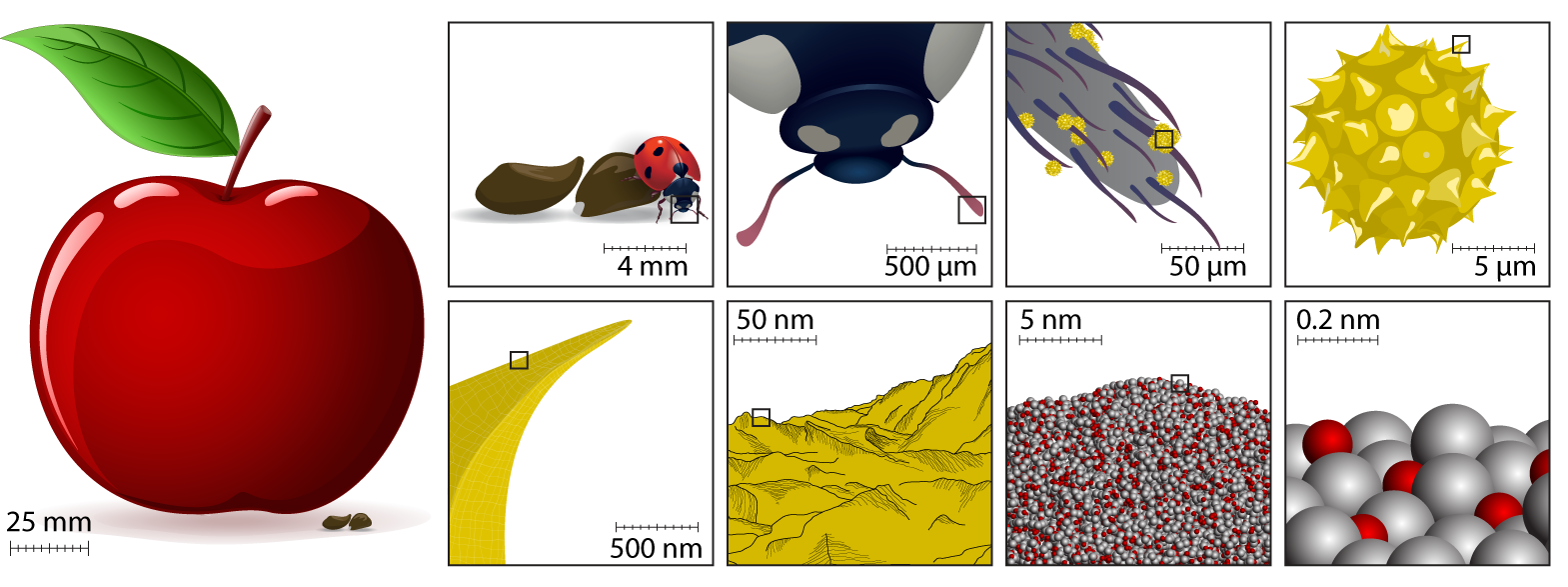
Another strength of this app is the quality of documentation and extra information available in the app and on the top bar of the website. I'm particularly impressed by the animated figure on "how big is an atom", which reminded me of the orders-of-ten approach. I would be lovely to see a similar explanation of the nano-second.
Disclaimer: I was made aware of this app by the developer, but I bought my own copy, and was not asked to write a review on the blog.