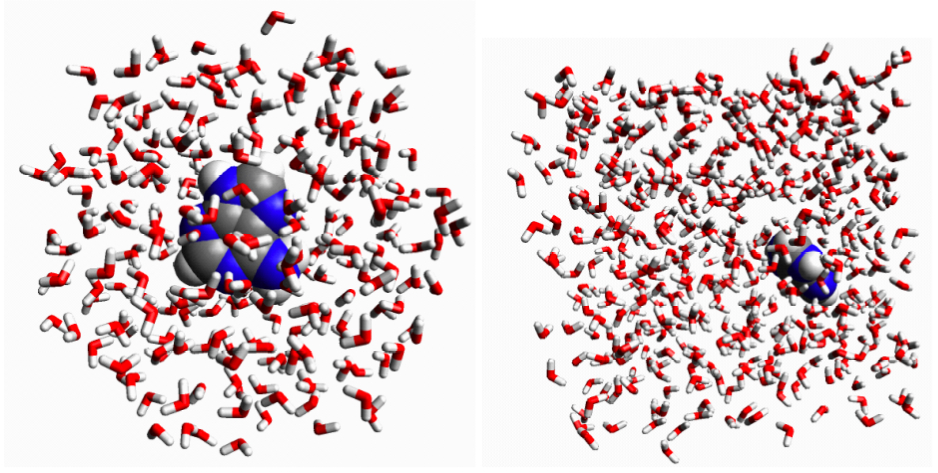
Figure 3.8. (a) Adenine (C5H5N5) in a liquid drop of 246 water molecules. (b) Adenine in a periodic box of 511 water molecules.
From Molecular Modeling Basics CRC Press, May 2010.
From Molecular Modeling Basics CRC Press, May 2010.
Conceptually, the simplest way of simulating a molecule in solution is to place it in the middle of a roughly spherical ball of water molecules (Figure 3.8a) and perform an molecular dynamics simulation.
One problem is this approach is that the drop would eventually evaporate if the simulation is run long enough. Another problem with the liquid drop model is that the water molecules at the surface of the drop do not behave like water molecules in liquid water.
Therefore, most explicit solvation simulations use periodic boundary conditions (Figure 3.9).
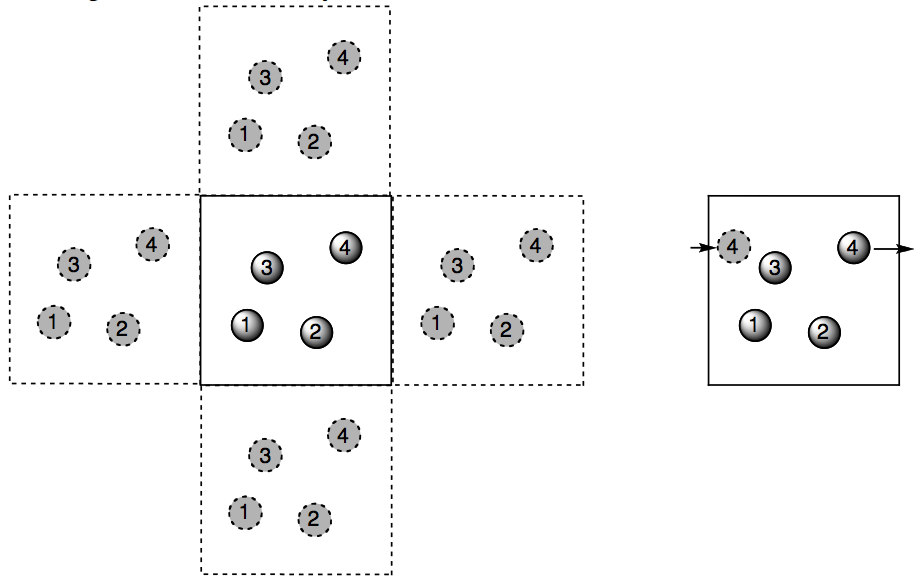
Figure 3.9. Sketch of periodic boundary in two dimensions: (a) The position of the particles in the central box are copied and placed in neighboring boxes. Figure 3.8b shows a cube from a real simulation. (b) When a molecule tries to leave the box during an MD simulation, it reappears at the opposite end of the box, so the number of particles in the central box stays constant.
From Molecular Modeling Basics CRC Press, May 2010.
And you can find an animated version of Figure 3.9 here (an example of where a movie really is worth 10,000 words).
From Molecular Modeling Basics CRC Press, May 2010.
You can find interactive versions of Figures 3.8a and 3.9b here (I am grateful to Kestutis Aidas for providing the coordinates).
Click on the picture for an interactive version

Click on the picture for an interactive version
And you can find an animated version of Figure 3.9 here (an example of where a movie really is worth 10,000 words).
The animation was made with Molecular Workbench (MW). You can play with the simulation here or you can download the MW file here (after you gave installed MW).



6 comments:
So here's a question -- I was just asked about implementing periodic boundary conditions for force fields in Avogadro.
Moving atoms across unit cells isn't hard to implement. Neither is it difficult to set up interactions with atoms in neighboring boxes.
My question: how many replica unit cells do you need (e.g., for proper electrostatics)? Is that something that needs to be a user-defined parameter?
Let me start by saying that I am not an MD expert (but I play one the blog :)).
It is my understanding that for non-charged molecules, one usually chooses a cutoff that is smaller than half the box size. In that case you only need one layer of replica unit cells (the minimum image convention).
For charged molecules or very polar molecules with large partial charges, particle mesh Ewald or similar techniques are needed for long-range interactions. I believe the minimum image can still be used here, but am not sure.
Tristan Youngs, who we met at the Daresbury meeting, may be a good person to ask for details. He may even donate code form Aten. Unfortunately the Aten web site (www.projectaten.org) is not functioning at the moment.
Hi Jan:
Nice to see you are using MW to make tutorial movies. I noticed that the MW link on your page points to workbench.concord.org, which may not be supported any more. Can you change the link to mw.concord.org?
Charles
PS: I cannot wait to see your new book. Can we pre-order it?
Charles, done!
I believe it is possible to pre-order the book on Amazon.com.
By the way, it may interest you that the MW-based tutorial movie I made on tunneling is by far the most watched screencast on the blog, with over 1200 views!
Jan, I had a new post in my blog about how to embed MW applets in blogspot.com, as a response to your comment about it.
Charles Xie
Charles, that's terrific. I can't wait to try it!
Jan
Post a Comment