This screencast shows
Molecular Workbench simulations I have made to illustrate mixing.
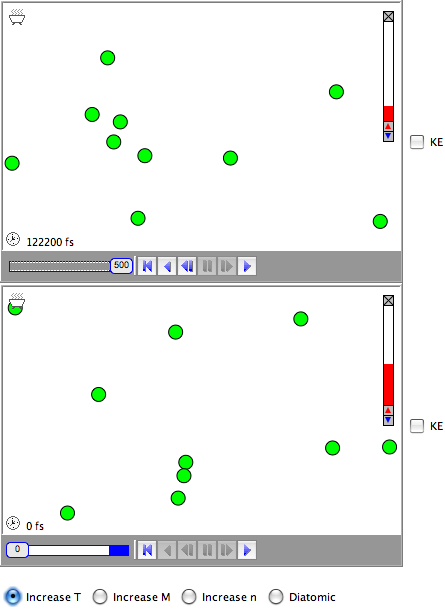
The first simulation illustrates the mixing of 2 ideal gases, which mix readily. Since the gas particles don't interact you can think of the mixing as each gas expanding to fill both containers independently of each other.
As I have shown in this simulation, the driving force for this expansion is an increase in entropy. Therefore, the driving force for mixing two ideal gasses is also purely entropic.
The second set of simulations illustrates the mixing of 2 liquids. Since they are liquids there must be attractive interactions between the atoms. If there were no interactions they would be (ideal) gasses. The strength of the interactions (and the temperature) determine whether they mix or not.
In the first liquid simulation, the attraction between two green atoms (
εGG), between two blue atoms (
εBB), and between a green and a blue atom (
εGB) are the same.
This means that a green atom doesn't care whether it is sitting next to a blue atom or another green atom. The net effect is that green and red atoms are equally likely to be on the right or left side of the container, and the liquids mix for the same reason as the ideal gasses mix: the driving force is purely entropic. That means the enthalpy of mixing is zero:
This is the definition of an
ideal mixture (or ideal solution). The two liquids will mix at any temperature.
In the second liquid simulation, the attraction between two blue atoms (
εBB) is stronger than between two green atoms (
εGG) and between a green and a blue atom (
εGB).
Note that the
ε's are negative: a smaller
ε means a stronger attraction. This means that the blue particles would rather be with other blue particles, i.e. the enthalpy increases if the particles are mixed.
(
z is the number of contacts between particles in solution, and
xG is the
mole fraction of green atoms). This is an example of a non-ideal mixture, where the definition for a non-ideal mixture is
Because Δ
mixH > 0 this non-ideal solution mixes spontaneously (i.e. Δ
mixG < 0) only for
Oil and water is a common example of such an non-ideal mixture: the oil-oil interactions are stronger than the oil-water and water-water interactions.
Salt and water is another example of on idea mixture, but here Δ
mixH < 0 so salt and water almost always mixes spontaneously. The interpretation is that the interactions between the salt ions and water is stronger than the average interaction between salt ions and between water molecules.
Implications and limitations
The definition of Δ
mixH in terms of the
ε's suggest that liquids should also mix if
which would be a more general definition of an ideal mixture.
This is tested in the third liquid simulation. As you can see the liquids mix more than in the second simulation, but not quite as much as in the first simulation. This is mostly because of the simulation runs only for 100 picoseconds, which is to short to mix fully. But another reason is that there is less space (on average) between the blue atoms compared to the green atoms, because the blue atoms attract each other more. The next effect is that the blue particles tend to stay together to lower the enthalpy. More mathematically,
z (the number of contacts between particles in solution) is not exactly the same for the blue and green particles so the interpretation of Δ
mixH in terms of the
ε's breaks down. The "safest" definition of an ideal mixture thus remains:
i.e. "like dissolves like".
Accessing the simulations
You can play around with the simulations
here and
here, or you can download the models
here and
here if you have
Molecular Workbench installed on your computer.
































{kind=link}