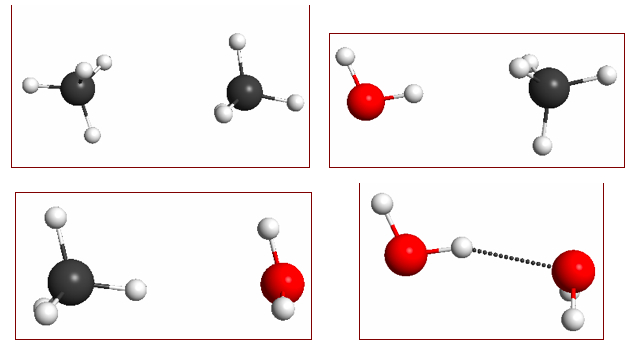
Figure 4.16. 0.002 au isodensity surface with superimposed molecular electrostatic potential for (a) methane using the methane density for the methane–water dimer and (b) free methane monomer. The maximum potential value, 0.01 au, is five times smaller than in previous figures to make the increase in negative charge visible. The black spheres in (a) denote the position of the three nuclei of water. The level of theory is M06/6-31+G(2d,p)//M06/6-31G(d).
Click on the picture for an interactive version.
Click here for a pop-up window
From Molecular Modeling Basics CRC Press, 2010
Click on the picture for an interactive version.
Click here for a pop-up window
From Molecular Modeling Basics CRC Press, 2010
In a previous post I showed how the difference in the strengths of interaction between methane, methane-water, and water dimer is due to their differences in polarity, which can be visualized using molecular electrostatic potentials (MEPs). Methane interacts stronger with water than with methane because of polarization, i.e. rearrangement of the methane electrons due to the polar water molecule that, to a first approximation, induces a dipole moment in methane.
The polarization of the methane density is not visible in the MEP of the methane–water dimer (Figure 4.15b) because the polarity of the water H atom dominates the MEP in that region. But if we remove the water molecule, the net increase in negative charge in the methane molecule where it interacts with the partially positive H atom of the water is apparent (Figure 4.16).
Making the figure
This is a not a plot one makes every day, so the process is a bit involved. The main trick is to construct the methane part of the density from the corresponding localized molecular orbitals, and then tricking GAMESS into printing a file with just those LMOs that MacMolPlt can read. The procedure is described in some detail Section 5.5 of the book, so here I just post the corresponding screencast.