Figure 4.10. 0.045 au isosurfaces of the four valence canonical MOs of NH3 computed using B3LYP/6-31G(d). A value of 0.045 au is chosen because it results in a 0.002 au isodensity surface when squared.
From Molecular Modeling Basics CRC Press, May 2010.
Click on the picture for an interactive version
From Molecular Modeling Basics CRC Press, May 2010.
Click on the picture for an interactive version
According to Valence Shell Electron Pair Repulsion (VSEPR) theory many bond angles involving elements such as C, N, and O are close to 109.5o because the four valence electron pairs that surround these atoms adopt a tetrahedral geometry to minimize repulsion. In the case of CH4 the H-C-H angle is exactly 109.5o because the repulsion between the four electron pairs in the C-H bonds are identical. NH3 has a lone pair that is fatter than a bond near the nucleus, so the lone pair-bond repulsion is slightly larger than the bond-bond repulsion. This results in a H-N-H angle of 107o, slightly smaller than 109.5o.
However, this is far from obvious when looking at the four valence MOs of NH3 (Figure 4.10) computed using B3LYP/6-31G(d). The reason is that that MOs are not unique, and that these MOs (which lead to a diagonal Fock matrix and are known as canonical MOs) are not the MOs where the electron pair repulsion is a minimum. Algorithms have been implemented that find a new set of MOs (localized MOs or LMOs) that represent a linear combination of canonical MOs for which the MO–MO repulsion is a minimum (Figure 4.11).

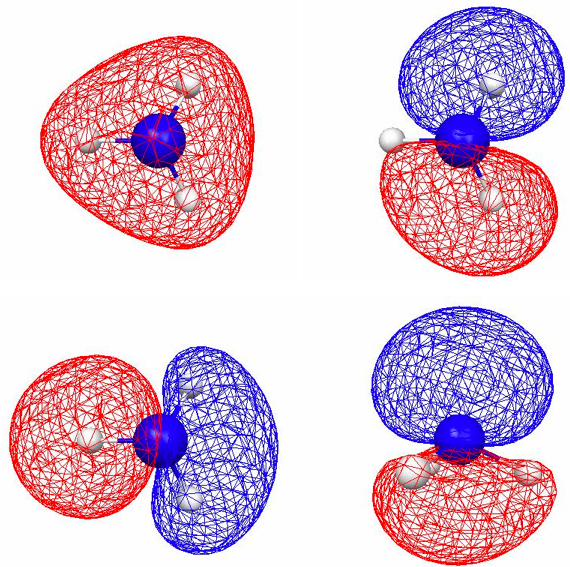
Figure 4.11. 0.045 au isosurfaces of the four valence localized MOs of NH3 computed using B3LYP/6-31G(d). There are three N–H bond LMOs [(a)–(c)] and one lone pair LMO [(d)].
From Molecular Modeling Basics CRC Press, May 2010.
Click on the picture for an interactive version
LMOs for which the inter-orbital repulsion is a minimum are called energy localized orbitals or Edmiston-Ruedenberg orbitals. Other popular choices are Foster-Boys and Pipek-Mezey LMOs, which use different localization criteria.
Using MacMolPlt
The screencast below shows how to compute Ruedenberg LMOs for ammonia using GAMESS (local=ruednbrg in the $contrl group), and how to display the LMOs, as well as the canonical MOs, in MacMolPlt. I also show how to identify the HOMO and LUMO canonical MOs in MacMolPlt.
Note that I specify a geometry optimization in the GAMESS input file. GAMESS will only compute the LMOs for the optimized geometry.
From Molecular Modeling Basics CRC Press, May 2010.
Click on the picture for an interactive version
LMOs for which the inter-orbital repulsion is a minimum are called energy localized orbitals or Edmiston-Ruedenberg orbitals. Other popular choices are Foster-Boys and Pipek-Mezey LMOs, which use different localization criteria.
Using MacMolPlt
The screencast below shows how to compute Ruedenberg LMOs for ammonia using GAMESS (local=ruednbrg in the $contrl group), and how to display the LMOs, as well as the canonical MOs, in MacMolPlt. I also show how to identify the HOMO and LUMO canonical MOs in MacMolPlt.
Note that I specify a geometry optimization in the GAMESS input file. GAMESS will only compute the LMOs for the optimized geometry.
Using Jmol
I use Jmol for the interactive figures and the scripts can be found here and here. Unlike the density and electrostatic potential, Jmol can generate its own grid data, so to display MO number 2 you simply use "mo 2" in the script. I use
mo 2; mo cutoff 0.045; mo fill nomesh; mo translucent 0.2to make it a little prettier.
Jmol cannot find the MOs in a GAMESS geometry optimization file, and Jmol only stores the LMOs if present. So to display the canonical MOs with Jmol you need a single point energy calculation (output file) and to display the LMOs with Jmol you need a single point energy calculation with local=ruednberg added (output file).



12 comments:
Jan,
Great post on MO's. Really appreciate the work.
NUChem
NUChem,
Very glad you liked it. Good to hear from you again.
Jan
Cool
I was just using Jmol to look at the MO's from a PC-GAMESS job on a steroid this morning. Jmol definitely draws MO's on steroids as it is so fast.
Thanks for making LMO's clear.
Bill
Bill, glad you found the post useful.
You raise a good point which I forgot to mention in the post: for large molecules, Jmol is much faster than MacMolPlt when plotting MOs.
Jan,
This is off topic, but I was wondering if you could suggest some programs that can do conformational analysis. I'm looking for a decent software package (ideally free but I know they don't exist :) that can do Monte Carlo conform searches on a molecule to find the global minimum and local minimums within a certain kcal energy above the global. Do you have any suggestions? Thanks a million.
NUChem
NUChem,
Avogadro can do this but the output is not ranked and you have do weed out duplicates yourself.
Marvin can also do this (under the tools menu) and the output is a bit nicer, but it is more work extracting the coordinates for each.
Both are free and work only with force field. Spartan can also do this at other levels of theory, but costs.
Hope this helps.
Jan
Jan,
I could use some help with Jmol. I have been working on a new website for students taking organic chemistry and have been trying to use jmol in the website design.
I have been successful on a few counts, but it seems that I keep running into issues when trying to use *.log files. I have been implementing jmol in a few ways (in the source code). Your coding has been very helpful, but not really knowing java or coding in general I have been doing my best to fake it via trial and error.
The website is brand new... in the "alpha" stages of development. so forgive the mess.
the redirect is www.tinyurl.com/liuorganic
a few unlisted test pages where I am having trouble are:
http://myweb.brooklyn.liu.edu/jgough/ChemResources/OrganicChem/Chapter2/test.html
http://myweb.brooklyn.liu.edu/jgough/jmol/test2.html
I can't seem to figure out how to:
1. load instructions from a file as you describe in the "Canonical and localized molecular orbitals" post.
2. most important, sometimes I can load a gamess .log file, but when i do in test.html (above) I can't figure out how to show orbitals. I really would like to be able show sp2 and sp3 hybridized orbitals.
Any help or guidance regarding scripting would be appreciated. I have blown about 6 hours trying different things thus far.... but I won't give up!!!
FYI - your blog and book were an important part of the inspiration behind the website.
Jonathan,
I got your comment by email, but for some reason it is not showing up here on the blog. I don't understand why.
Anyway, I will look at your questions and get back to you. It will probably take me a little while.
Jonathan,
Here is how I create the interactive figure 4.10 in the post:
The insertJmol script loads the file fig4-10.xyz.
This file loads the script file fig4-10.spt, which loads the GAMESS output file, and displays the MOs.
Hope this helps. If not let me know. Don't give up!
Dear Jan,
Can Ruedenberg method print virtual orbitals like NBO analysis?
Best Regards,
Lucas.
No, not as far as I know
Thank you, Jan.
Hugs,
Lucas
Post a Comment