[source]
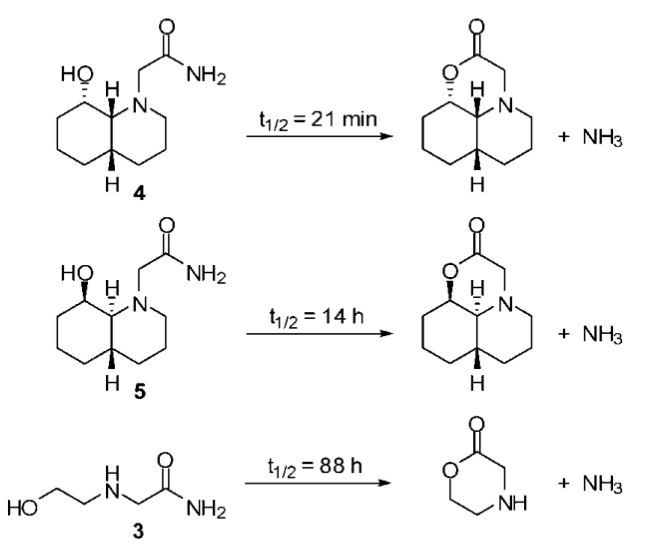
In a previous post I showed how to find a TS for an Sn2 reaction involving a small molecule (it's a good idea to look at this post now if you haven't seen it before, and check out this post as well).In this and future posts I discuss the steps involved in finding a TS for a reaction involving complicated molecules shown in the figure above (I have shown how to build some of the reactants in a previous post).
Say you want to find the TS for the hydrolysis of 4 and 5 at a relatively high level of theory. My advice is to start by finding the TS at a low level of theory (PM3) for a smaller representative reaction (hydrolysis of 3). This is the subject of today's post.
The basic procedure is the same as in the previous post:
1. I build a TS-like structure by constraining a key distance during an energy minimization.
2. I check whether the optimized geometry has an imaginary frequency that looks like the reaction coordinate
3. If it does, I use it as a starting point for the TS search.
In the previous example, one bond broke as the other one formed, so I simply forced one bond (the C-F bond) to form by constraining it to 2.0 Å and the optimization took care of the rest.
In the hydrolysis of 3, two bonds have to break (the C-N and O-H bonds) as two other bonds form (the C-O and N-H bonds), so what to constrain? In general I have found it a good idea to try to construct an intermediate structure, such as "3TSa" shown below.

Why do I pick 2.0 Å? Well, you have to start somewhere and this is a nice round number (for bonds involving 3rd row atoms I would use 2.5 Å). As you see in the screencast below, 2.0 Å doesn't work but the TS search tells me how to change it, i.e. increase it to make it more reactant like as the TS search goes to products.
As you saw in the screencast I have pre-typed some text with keywords that I use again and again, and simply paste the text in the input file and alter it when necessary. For the constrained optimization the text looks like this:
$contrl runtyp=optimize $end
$basis gbasis=pm3 $end
$statpt opttol=0.0005 nstep=50 hssend=.t. $end
$force nvib=2 $end
$contrl nzvar=1 $end
$zmat dlc=.t. auto=.t. ifzmat(1)=1,x,x fvalue(1)=2.00 $end
and for the TS search the text looks like this.
$contrl runtyp=sadpoint $end
$basis gbasis=pm3 $end
$statpt opttol=0.0005 nstep=50 $end
$statpt hess=calc ihrep=5 hssend=.t. $end
$force nvib=2 $end
$contrl nzvar=1 $end
$zmat dlc=.t. auto=.t. $end
You might also notice that I am being a "CPU slob" in the sense that I redo some computations I've already done. For example, I compute the frequencies for the optimized constrained geometry twice: at the end of the optimization and at the beginning of the TS search. That is because I am using PM3, which is such an efficient method that the computer can re-compute the freqeuncies in less time than it takes me to transfer the $HESS group to the TS search input file (as shown in a previous screencast)! Similarly, when I find that 50 steps is insufficient and just restart with nstep=100 steps, which recomputes the first 50 steps. Good thing sloth is not a deadly sin.
Another advantage of constructing a hypothetical intermediate in this case is that you realize (hopefully) that the TS contains a chiral center, even though the reactants and products are achiral. So you have to look for two TSs, to see which one is lowest in energy.
The next screencast shows how I find 3TSb, which is made easier by the fact that I have a good idea of what the constraint value should be (2.32 Å) from the 3TSa geometry I found previously.



4 comments:
Hi,
Can you clarify this fragment:
"The reason for choosing the C-O bond is that I assumed that the (potential) energy of 3 is lower than the products (because of ring strain) so the TS should be more reactant-like according to Hammond's postulate (you can of course check this computationally)."
If energy of 3 is lower than energy of the product, that means that the reaction is endothermic (deltaH>0). Hammond postulate says that for endothermic reaction TS is a product-like. I do not understand why you say that this is the reactant-like TS. :-(
Very nice blog :-)
rac
That was a typo, which I have now corrected. Thanks very much for catching that.
In hindsight, the Hammond postulate was not a great argument for choosing the CO bond. But it is definitely the right choice, when one looks at the TS.
Just watching over a few of these older screencasts, it is amazingly helpful to see how you use the tools available (and to get ideas on how to improve them further). Thanks for taking the time to put these up (and in a way where they remain available). Just learning more about transition states and GAMESS, in the process got some great new ideas for interface too.
I am very happy to hear you find the screencasts useful, especially if they inspire new ideas. Thanks very much for letting me know!
It's actually funny, coming from you. Your screencasts of Avogadro were actually what inspired me to get me started in the "screencast business" in the first place, and alerted me to blip.tv.
Post a Comment