In a previous post I showed how to compute an electrostatic potential map superimposed on the 0.002 isodensity surface of a molecule based on data computed using quantum chemical methods such as RHF/6-31G(d).
Previous posts (such as this one) have demonstrated that the van der Waals surface is a reasonable substitute for the 0.002 isodensity surface. Similarly, the electrostatic potential due to atomic charges can be a reasonable substitute for the electrostatic potential due to the electronic density.
The screencast above shows how to make such electrostatic potential maps using Avogadro and Jmol.
Avogadro uses an empirical method to determine the atomic charges (an integral part of the MMFF force field). It is possible to change the surface using the so-called "Iso Value" but I could not find any documentation on how that actually works. An Iso Value of 0 seems to correspond to the van der Waals sphere surface. It is currently not possible to alter the color range as far as I can see.
Jmol is not able to determine the charges, but the information can be transferred from Avogadro by saving a mol2 file. The electrostatic potential option in the Jmol menu corresponds to the following set of commands (as far as I can determine):
isosurface solvent color range -0.05 0.05 map mep color isosurface translucent 0.5and this set of commands can thus be used to control the color range and the nature of the surface. The default surface is a solvent accessible surface, which is slightly larger than the van der Waals surface.
A static and interactive version of the Avogadro and Jmol electrostatic potential map, respectively, can be found here.
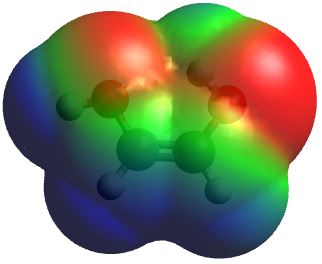
Electrostatic potential map made with Avogadro
Click on the picture for an interactive version made with Jmol
Click on the picture for an interactive version made with Jmol
Related blogpost: The polarity and solvation option in MolCalc



4 comments:
Actually, the charges currently come from the Gasteiger-Marsili model, not MMFF94. It's probably better to use MMFF94 charges, but that's not what happens in version 1.0.
OK, thanks Geoff. It seems that it is also the Gasteiger-Marsili charges that are displayed with the label option? I agree that it would be better to display the FF charges (or at least have the option).
Any thoughts on how the Iso Value works? i.e. exactly how does it affect the surface?
I want to know is it possible to calculate only electronic contribution to the electrostatic potential ( i.e., not the total ) on a particular atomic coordinate in GAMESS?how to do it?
Have a look a $elpot. That might do it.
Post a Comment